
Studies on genome and genetic resources in Brassicaceae crops
We have a long history on the study of classical genome analysis of Brassicaceae species in our laboratory. The scientific name of Chinese cabbage and turnip is Brassica rapa L., that of cabbage and broccoli being Brassica oleracea L., that of rapeseed being Brassica napus L., and that of mustard being Brassica juncea Czern. Genome relationships between six species including them were revealed in Japan by observation of chromosome pairing in pollen mother cells at meiosis, and these relationships are known as “triangle of U”. Professor Mizushima, the first professor of this laboratory, greatly contributed to this study, and further studied genome relationships of many species related to Brassica (Mizushima 1980 Brassica crops and wild-allies pp 89-106).
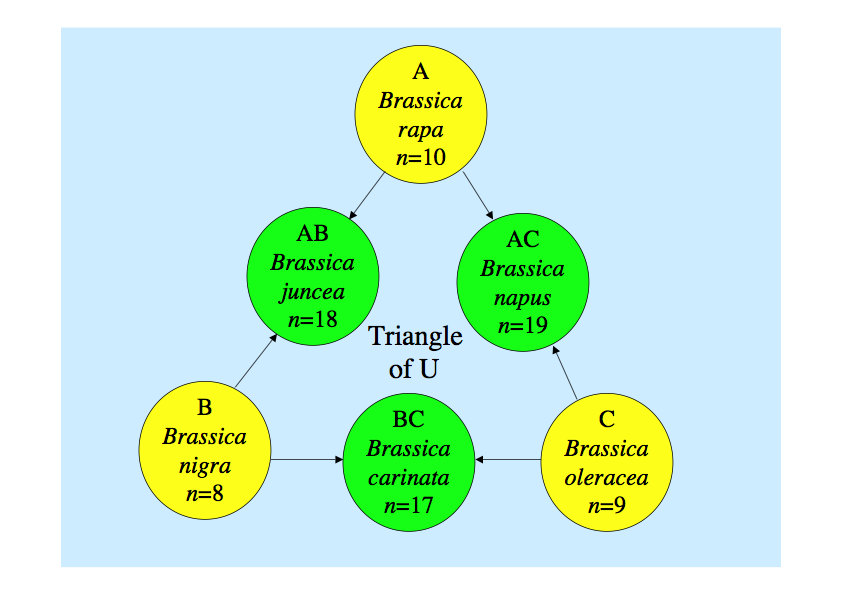
“Triangle of U” showing genome relationships between species in Brassica
At present, “genome” is used as a term meaning the total genetic information held by an organism, and “genome study” means determination of nucleotide sequences of all chromosomes from end to end. For determination of nucleotide sequences of the genomes and identification of genes controlling various trait, construction of linkage maps of DNA markers are indispensable. To identify genes responsible for various traits important in breeding of Chinese cabbage (B. rapa), cabbage (B. oleracea), and radish (Raphanus sativus), we produced many DNA markers and constructed linkage maps. In this study, we developed mainly single nucleotide polymorphism (SNP) markers that can be analyzed by our original technique for SNP analysis, dot-blot-SNP technique (Shirasawa et al. 2006 Theor Appl Genet 113, 147-155 Please see “Development of breeding techniques using SNP analysis of genes”.). Using these SNP markers, we have detected quantitative trait loci (QTLs) for glucosinolate content in radish root (Zou et al. 2013 Plos One 8, e53541), flowering time of B. rapa (Li et al. 2009 DNA Res 16, 311-323), variation of leaf hair in B. rapa and R. sativus (Li et al. 2013 Theor Appl Genet 126, 1227-1236), and black rot resistance in B. oleracea (Kifuji et al. 2013 Euphytica 190, 289-295), and identified or inferred genes responsible for these traits.
Although B. rapa is closely related to B. oleracea, chromosome numbers of these species are different, being n = 10 in B. rapa and n = 9 in B. oleracea. Comparison of linkage maps of genes between these species reveals that the difference of chromosome numbers is due not to simple cleavage of one chromosome but to complicated genome rearrangements. However, it was found that chromosome 1 and 2 are similar between these two species and chromosome 9 in B. rapa is similar to chromosome 8 in B. oleracea (Ashutosh et al. 2012 Mol Breed 30, 1781-1792). Similarities of linkage maps between B. rapa and R. sativus are more complicated, reflecting more distant relationship of R. sativus than B. oleracea to B. rapa. In the past, homology of chromosomes has been analyzed by chromosome pairing at meiosis, but can be analyzed in detail at molecular level, now. Reading of the whole genome nucleotide sequences of these species enables elucidation of relationships of genomes of different species and inference of the process of genome evolution.
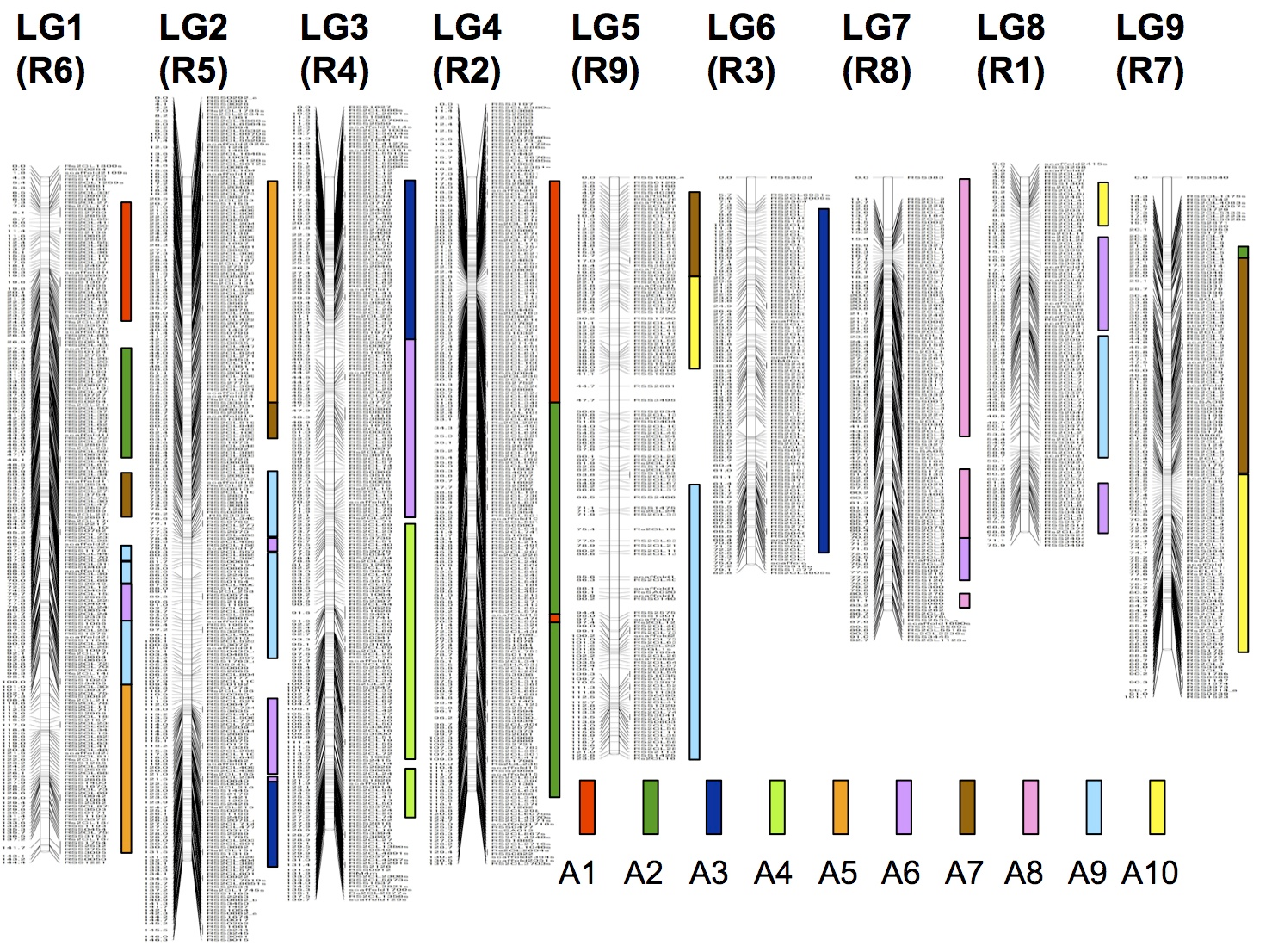
Figure. A linkage map of genes in R. sativus and comparison with that in B. rapa (bars to the right)
In 2008, we started “Daikon” genome project supported by the Program for Promotion of Basic and Applied Researches for Innovations in Bio-oriented Industry (BRAIN), Japan. We constructed a linkage map of ca. 1,000 SNP markers of genes of R. sativus, and combined with a linkage map of SSR markers, which has been constructed by Kazusa DNA Research Institute. To the integrated linkage map, SNP markers of genomic sequences were added, and finally a high-density linkage map with 2,550 DNA markers was produced. A large number of nucleotide sequences of genomic DNA of R. sativus cv. ‘Aokubi’ were obtained by next generation sequencers, and end sequences of BAC (Bacterial Artificial Chromosome) clones were determined. Cooperating with members of Kazusa DNA Research Institute, we determined ca. 400 Mb sequences of the R. sativus genome (76% of the total genome) (Kitashiba et al. 2014 DNA Res doi:10.1093). These genome sequences are published at the Website of ‘Raphanus sativus Genome Data Base’ (http://radish.kazusa.or.jp). Furthermore, SNPs in more than 1,000 genes between eight cultivars were identified by cooperating with Iwate University, National Institute of Vegetable Research, and Takii Seed Co. Ltd. (Kitashiba et al. 2014 DNA Res doi:10.1093).
Since this laboratory has a long history of genetic and breeding studies of Brassicaceae crops, we have a large number of species and lines in the Tribe Brassiceae, which are useful in the study of genetics and breeding of Brassicaceae crops, and provide seeds of them to researchers in the world (Please see Tohoku Univ. Brassica Seed Bank.). In these Brassicaceae genetic resources, there are rare species, which are not known even by specialists of Brassicaceae crops. To avoid confusion of species, we developed simple methods for identification of species in Brassiceae (Tonosaki and Nishio 2010 Plant Cell Rep 29, 1179-1186, Tonosaki et al. 2013 Mol Breed 31, 419-428). For effective use of these genetic resources, phylogenetic relationships of species were revealed by nucleotide sequence analysis of a common single copy gene (Inaba and Nishio 2002 Theor Appl Genet 105, 1159-1165). These genetic resources have scarcity value and well-known by a community of Brassicaceae crop specialists in the world.→"Tohoku Univ. Brassica Seed Bank"